
Genetics of male infertility
Our Male Infertility Specialist:
It is clear that a significant proportion of infertile male with azoospermia and severe oligospermia have a genetic etiology for reproductive failure. While recent advances in assisted reproductive technologies make possible and practical for many infertile men with severe male factor infertility to father children, they also raised concerns about passing on genetic abnormalities to the offspring of these men. Intracytoplasmic sperm injection (ICSI) is the most invasive technique for assisted reproduction. ICSI bypasses all the physiological mechanisms related to fertilization as well as all protective barriers against sperm with genetic defects and allow even altered spermatozoon to fertilize an oocyte. Since infertile patients with non-obstructive azoospermia are able to achieve pregnancy with surgically retrieved testicular sperm, ICSI carries risk of transmitting both genetically determined diseases and genetically determined infertility. It is imperative for the clinicians involved in the treatment of these couples to initiate genetic evaluation and counseling prior to any therapeutic procedures.
All human nucleated diploid cells contain 22 pairs of autosomes and 1 pair of sex chromosomes, for a total of 23 pairs, which provides the normal 46, XX (female) and 46,XY (male) chromosomal configuration. It is important to recognize that the only source of Y chromosome is paternal.
It is estimated that human genome contains 50,000-100,000 genes that encodes proteins.
Cystic Fibrosis
Cystic Fibrosis (CF) gene mutation have been detected in men with congenital absence of the vas deferens, but with no other manifestation of CF. CF is the most common fatal autosomal recessive disorders in the Caucasian population with the incidence approximately 1 in 2400 live births. Carrier frequency is higher at 1 in 25 persons in population of Northern European descent. The CF gene was identified and cloned in 1989 and has been named the Cystic fibrosis transmembrane conductance regulator gene (CFTR). Genetic studies revealed that 50-83% of patients with Congenital Bilateral Absence of the Vas Deferens(CBAVD) have at least one known CFTR gene mutation and that approximately 10% have two known CFTR mutations. Most men with CBAVD have only one detectable gene mutation or variant. The most commonly found mutations are D 508 (70%), 5T, R117H, R75Q, G542X, N1303, W1282X, G551A, and R347H. In those men with mutations identified in both CFTR alleles at least one mutation is mild. More than 95% of CF men have abnormalities in the structures derived from Wolffian duct. Absence of the vas deferens may be the only presenting clinical sign in these patients. Corpus and cauda epididymis may be present, absent or atrophic, but caput epididymis is present invariably. It has been speculated that the longer the epididymal remnant, the higher the success rate of sperm aspiration. CBAVD may represent mild from of CF. Up to 82% of patients with CBAVD have at least one detectable CF mutation. Congenital bilateral absence of the vas deferens is found in 2% of men presenting with infertility. It is a common cause of azoospermia associated with low semen volume and acidic pH. Spermatogenesis is usually intact, therefore testicular biopsy is presently not recommended in these patients. Sufficient number of spermatozoa could be usually retrieved from the obstructed epididymis or its remnant.
If a man with CBAVD is contemplating microsurgical epididymal sperm aspiration with subsequent ICSI, the complete spousal CF mutation analysis is mandatory to define the risk of passing along CF or CBAVD. If spouse is negative for the common mutation, her CF carrier risk is about 0.4%. A risk for this couple to have a child with CF genotype is no more than 0.2%. If the wife is a carrier, then the chances for child to have CF genotype is 50%.
Congenital Unilateral Absence of the Vas Deferens(CUAVD) affects 0.5-1% of the general population and only rarely presents with infertility. CFTR gene mutations have been reported in 43% of men with unilateral absence of the vas deferens and 47% of otherwise healthy men with idiopathic epididymal obstruction.
Urogenital anomalies in patients with the absent vas deferens.
Renal agenesis was found in 11% of patients with CBAVD and 26%of patients with CUAVD
Seminal vesicles were found in 55% of patients with CBAVD. Ipsilateral seminal vesicle was present in only 14% of patients with CUAVD.
Karyotype evaluation
The most common karyotype abnormality in men with severe male factor infertility is Klinefelter's syndrome, affecting 7-13% of azoospermic men. Prevalence of Klinefelter's syndrome is estimated at about 1 in 600 live-born males. The classic triad includes small firm testes, gynecomastia and azoospermia. 90% of all patients have 46,XXY Karyotype, and 10% are mosaic with Karyotype 46XY/47,XXY. Sperm retrieval is possible in these patients despite severe histological changes (hyalinization of the seminiferous tubules and hyperplasia of the Leydig cells) with subsequent fertilization and pregnancy.
It is also suggested that almost one in twenty oligospermic men had an abnormal Karyotype (mainly Robertsonian and reciprocal translocations). Karyotyping should therefore be regarded as a mandatory part of the pre-treatment screening process for all men referred for ICSI.
Y-chromosome Microdeletions
Tiepolo and Zufardi in 1976 found the deletion of distal end of the long arm of the Y chromosome. This region includes the azoospermia factor (AZF) locus, which contains a gene or genes, required for spermatogenesis. The AZF locus has been mapped to deletion interval 5-7, a region in band q11.23 of the Y chromosome that contains 5 million base pairs. There are three regions in deletion interval 5 and 6 where most deletions occur in infertile male, designated as AZFa, AZFb, and AZFc. Each of these regions may be associated with a particular testicular histology In one study microdeletions in the Y-chromosome were found in 7% of unselected group of infertile men which suggests that Y-chromosome microdeletions constitute the second most common specific cause of male infertility.
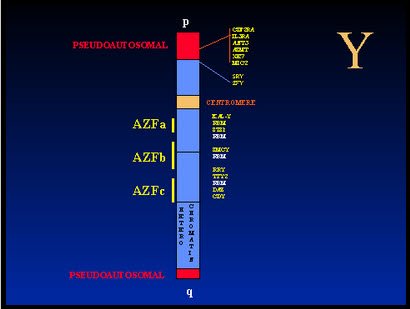
The AZF genes have not been identified yet. Two candidate gene families were cloned presently from deletion interval 6: RBM (RNA-Binding Motif) and DAZ (Deleted In Azoospermia). DAZ genes are expressed exclusively in the test. DAZ proteins seem to be present in late spermatids and in the tails of mature spermatozoa Absence of detectable deletions in normal fertile men suggests cause-and effect relationship between the deletions and infertility. Nevertheless, it was shown that some men with microdeletions may produce sperm in the ejaculate. Also, azoospermic men with deletion of the DAZ gene cluster occasionally are capable of completing spermatogenesis. It was speculated that AZFc microdeletions are compatible with formation of small numbers of normal spermatozoa. When retrieved testicular sperm were used for assisted reproduction (IVF/ICSI), normal embryonic development and pregnancy have been documented. The functional role of the Y-chromosome genes in spermatogenesis remained to be identified. It is suggested that Y-chromosome microdeletions occur during DNA replication. Among the men with azoospermia or severe oligospermia 16% had deletions. In men with azoospermia only the frequency of deletions increased to 23%.
Y-chromosome microdeletions are relatively frequent and their frequency increases with the severity of the spermatogenic defect. Therefore, testing for Y-chromosome microdeletions is indicated in men with azoospermia and severe oligospermia, and in patients considering assisted reproduction.
The Y-chromosome microdeletions cannot be predicted on the basis of clinical findings or the results of semen analysis. The testing for Y-chromosome microdeletions involves extraction of DNA from white blood cells and amplification of specific region of Y-chromosome in PCR containing 5-8 primer pairs. Each primer pair amplifies a specific region of Y-chromosome (a sequence-tagged site).
Y-chromosome abnormalities will be passed on to any male child who is produced after the assisted reproduction. Since men with these genetic disorders are usually perfectly healthy, apart from infertility, it is uncertain if any other medical conditions will be present in the offsprings with Y chromosome microdeletions.
Genetic counseling is important prior to treatment. The detection for Y-chromosome microdeletions may provide the diagnosis for infertility and allow physician to direct patients to assisted reproduction or adoption. Since sons of men with Y-chromosome microdeletions conceived with assisted reproduction will be infertile, child's male factor infertility needs to be discussed.
Present data do not suggest increased risk of genetically transmitted diseases in children born after ICSI. Major malformations were found in a range of 2.4%, which is comparable to figures known from children born after IVF or natural conception. Nevertheless, patients need to be counseled before any treatment on the basis of the available data about the risk of transmitted chromosomal defects, de-novo chromosomal aberrations and fertility problems to the offspring.